Neurobiologia de la esquizofrenia
Neurotransmisión y
circuitos en Esquizofrenia
El mecanismo
neuroquímico de la Esquizofrenia:
Hipótesis
Dopaminérgica
Esta hipótesis sostiene que los síntomas de la Esquizofrenia se deben a un exceso de dopamina o a una elevada sensibilidad a este neurotransmisor (Matthysse, 1974). Se formuló tras el descubrimiento de que los antipsicóticos efectivos en la Esquizofrenia eran antagonistas de los receptores dopaminérgicos (Carlsson y Lindqvist, 1963) y tras la observación de que los agentes liberadores de dopamina podían producir síntomas psicóticos (Rotrosen et al., 1979; Thompson et al., 2004; Lieberman et al., 1987).
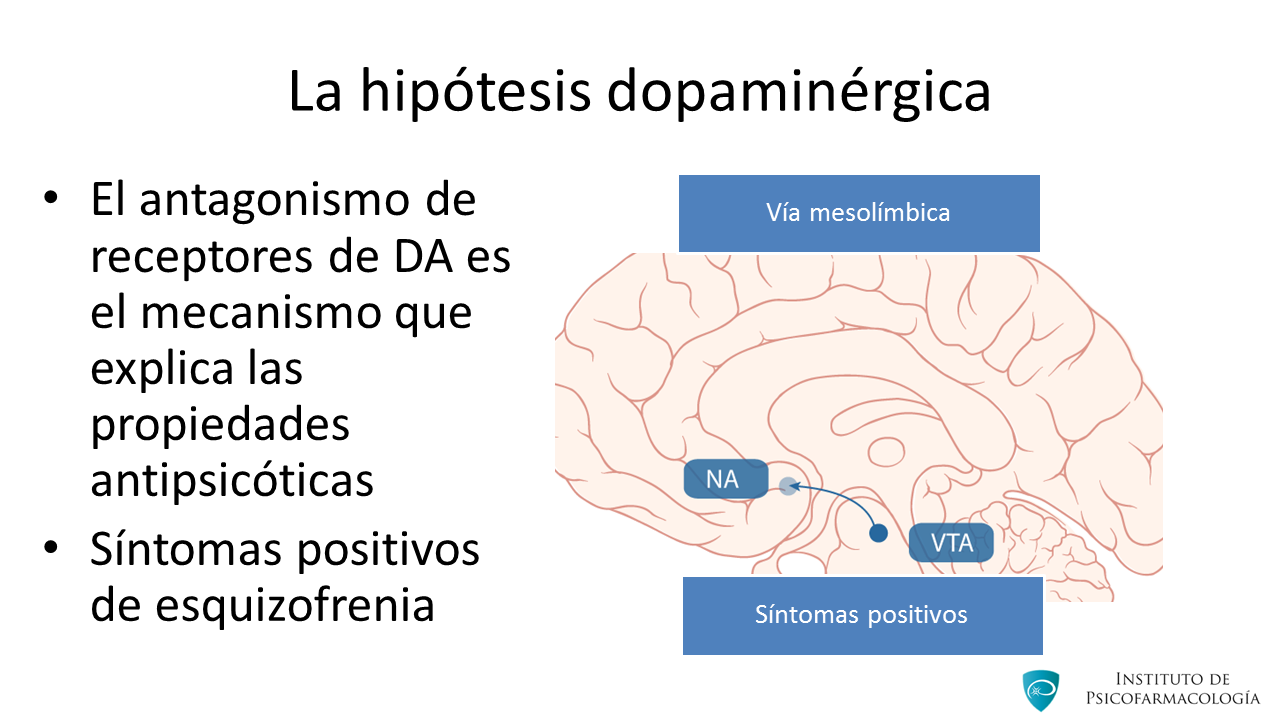
Las principales vías dopaminérgicas cerebrales que nos interesa señalar son:
Mesolímbica: Proyecta desde el área tegmental ventral del mesencéfalo a ciertas áreas límbicas, como el núcleo accumbens, que forma parte del circuito de recompensa. Teóricamente la hiperactividad dopaminérgica de esta vía explicaría la producción de los síntomas positivos en las psicosis.
Además, este circuito es importante para la regulación de las respuestas emocionales, la motivación, el placer y la recompensa, por lo que una disfunción a este nivel, podría explicar parte de los síntomas negativos observados en la Esquizofrenia.
En este caso, existiría un déficit en la función dopaminérgica. Quizás, la mayor incidencia de abuso de sustancias en la Esquizofrenia, podría explicarse como un intento de potenciar la función deficitaria de este sistema de recompensa o centro del placer mesolímbico (Grace, 1993; Grace, 1991a).
Por otro lado, la hiperactividad de las neuronas dopaminérgicas de esta vía puede desempeñar un papel en las conductas agresivas y hostiles de la Esquizofrenia, sobre todo si se asocia a un control serotoninérgico errático.
Mesocortical: Proyecta desde el área tegmental ventral a córtex prefrontal ventromedial
y dorsolateral.
Los haces que conectan con el córtex ventromedial, se han relacionado con funciones de regulación de emociones y afectividad, por lo que, un déficit dopaminérgico en esta vía podría explicar parte de los síntomas negativos y afectivos observados en la Esquizofrenia.
Por otro
lado, los haces que proyectan al cortex dorsolateral se relacionan con la
regulación de funciones cognitivas, por lo que algunos de los síntomas
negativos y cognitivos de la Esquizofrenia pueden ser debidos a un déficit de actividad
dopaminérgica a este nivel (Cropley et al., 2006).

Nigroestriada: Proyecta desde la sustancia negra del troncoencéfalo a los ganglios basales o estriado.
Esta vía
forma parte del sistema extrapiramidal y desempeña un papel clave en el control
de los movimientos motores. En la Esquizofrenia no tratada, esta vía puede
estar relativamente preservada. Sin embargo, las sustancias que bloquean los
receptores de dopamina D2 en esta vía, reproducen trastornos de movimiento como
la enfermedad de Parkinson (con temblor, rigidez y acinesia/bradicinesia),
acatisia y distonía, provocados por la deficiencia de dopamina a este nivel.
Cuando la dopamina está en exceso en esta vía, se producen movimientos
hipercinéticos como corea, tics o discinesias. Un ejemplo sería la discinesia
tardía inducida por neurolépticos que puede aparecer por el bloqueo crónico de
estos receptores en esta vía nigroestriada.

Tuberoinfundibular: Constituida por las neuronas que proyectan desde el hipotálamo a la hipófisis anterior, mediando en funciones neuroendocrinas.
Regula la secreción de prolactina a la circulación sanguínea inhibiendo su liberación. Al recibir tratamiento con fármacos que bloquean los receptores dopaminérgicos D2 en esta vía se elevan los niveles de prolactina, pudiendo surgir efectos secundarios (galactorrea, amenorrea y disfunción sexual). En pacientes con Esquizofrenia que no reciben tratamiento antipsicótico se considera normal el funcionamiento de esta vía.
La
hipótesis con más aceptación conceptual ha sido la dopaminérgica, a pesar de lo
cual, el grueso de los datos que la apoyan son indirectos (efecto psicotizante de
sustancias dopaminérgicas y efecto antipsicótico de fármacos
antidopaminérgicos) (Keshavan et al., 2008).

En la
versión revisada de la hipótesis dopaminérgica, se propone que la Esquizofrenia
se asociaría a una disregulación en la transmisión dopaminérgica: por un lado
se observaría
una hiperfunción dopaminérgica subcortical en las proyecciones mesolímbicas, que resultaría en la hiperestimulación de los receptores D2 con aparición de sintomatología positiva. Por otro lado, una hipofunción en las proyecciones dopaminérgicas mesocorticales al córtex prefrontal, que resulta en la hipoestimulación de los receptores D1 con la consecuente sintomatología negativa, afectiva y cognitiva.
Sin embargo, las vías dopaminérgicas nigroestriada y tuberoinfundibular permanecerían relativamente preservadas (Davis et al., 1991; Laruelle et al., 2003).
Para poder explicar
la complejidad de la Esquizofrenia se han propuesto teorías basadas en la
alteración del sistema dopaminérgico donde pueden tener cabida alteraciones en
otros sistemas de neurotransmisión. En este contexto se desarrolló la teoría de
la constricción de los límites de tolerancia a la dopamina, donde se plantea la
coexistencia de situaciones de hiperdopaminergia e hipodopaminergia relativa
por una alteración de la modulación de la actividad de este neurotransmisor
(Ashcroft et al., 1981; Grace, 1991b).
Esta modulación
dependería de otros neurotransmisores y de la interacción de los diferentes
receptores dopaminérgicos, existiendo mecanismos moduladores de compensación homeostática,
que implican disminución o aumento de receptores dopaminérgicos en función de
la concentración existente de dopamina en la hendidura sináptica.
De este modo,
cuando existe un exceso de concentración de dopamina en la hendidura sináptica,
se produce una disminución de receptores y a la inversa. En condiciones
normales, en respuesta a estímulos como el estrés, el disparo neuronal de dopamina
se produce de manera fásica, inmediata, siendo ésta rápidamente reincorporada
sin dar lugar a la mediación del mecanismo de homeostasis descrito.
Sin embargo, en
reposo la dopamina se libera de manera tónica a la hendidura sináptica,
mantenida más tiempo, dando lugar a la activación de los mecanismos de
compensación homeostática reguladores de la densidad de receptores. Esta
liberación tónica de dopamina se mantendría gracias a la actividad de la
corteza cerebral, que a través de proyecciones glutamatérgicas
corticosubcorticales conse-guiría un tono dopaminérgico adecuado (Grace, 1991b).
La disregulación
dopaminérgica subcortical observada en la Esquizofrenia podría ser secundaria al
fracaso de la corteza prefrontal (Weinberger y Lipska, 1995; Grace, 1991b;
Lewis y Levitt, 2002).
Así, Carlsson
describe un modelo en el que la corteza prefrontal modularía la actividad
cerebral del cerebro medio mediante una vía activadora, constituida por proyecciones
glutamatérgicas hacia las neuronas dopaminérgicas; y otra vía inhibitoria mediante
proyecciones glutamatérgicas hacia las interneuronas gabaérgicas (Carlsson y
Lindqvist, 1963).
Según el modelo de
Grace, en la Esquizofrenia existiría una hipoglutamatergia corticosubcortical, con
lo que la liberación tónica de dopamina estaría disminuida, disminuyendo su
concentración en la hendidura sináptica.
Consecuentemente a
esta disminución, se activarían mecanismos homeostáticos de hipersensibilidad
dopaminérgica, generando una hiperactivación dopaminérgica postsináptica en respuesta
a una actividad dopaminérgica fásica (Grace, 1991).

Hipótesis Glutamatérgica
Los mecanismos
propuestos para explicar la mediación del glutamato en la Esquizofrenia
encuentran su fundamento en la neurotoxicidad inducida por este neurotransmisor
y su interacción con la dopamina (Coyle, 2006).
Por tanto, los
antagonistas NMDA ocasionan una disminución de la excitación tónica sobre las
neuronas GABAérgicas, con lo que liberan la acción de las neuronas excitadoras.
Esta pérdida de modulación (inhibición) puede ser responsable del estado
psicótico que se induce (NAP), así como de la citotoxicidad (NAN).
Al considerar la
interacción entre las células glutamatérgicas por medio del receptor NMDA con
las células GABAérgicas, se plantea que una disminución (o ausencia congénita)
de estas últimas células puede desencadenar estados psicóticos y degeneración
neuronal.
Para simplificar la
explicación anterior, se considerará el tálamo. Este es un punto de relevo
aferente y eferente de la corteza cerebral.
Las neuronas
tálamo-corticales son glutamatérgicas y dan colaterales al núcleo reticular que
es GABAérgico, el cual manda proyecciones inhibitorias a las células
tálamo-corticales, con lo que forma un circuito de retroalimentación
inhibitorio. La pérdida de las células reticulares ocasiona una liberación de
este circuito excitador. Esta falta de inhibición se volverá más evidente
cuando termina la mielinización, la cual se da al inicio de la adolescencia, lo
que resulta en psicosis y daño por excitotoxicidad.
El glutamato puede
actuar también a través de receptores no-NMDA, por lo que su acción es
heterogénea al igual que lo es la de la dopamina. Al considerar que una
hipoactivación de los receptores NMDA ocasiona una liberación GABAérgica sobre
neuronas glutamatérgicas, se puede entender la presencia de cierta heterogeneidad
en la concentración glutamatérgica en el SNC.
En el tálamo, la
“hipofunción” del núcleo reticular puede ser ocasionada por la ausencia de
células GABAérgicas, así como por un estado hipoglutamatérgico, los cuales
inducen un incremento en el tono glutamatérgico tálamo-cortical.
Es interesante
señalar que el efecto de NAN ocasiona en la rata cambios patológicos en las
mismas regiones cerebrales que las encontradas en la esquizofrenia.
Estos cambios se han reportado en la corteza del cíngulo, hipocampo, giro parahipocámpico y corteza entorrinal. antipsicóticos. Deberse dopamina surte efecto glutama-térgica. Manifiesta en la regulación glutamatérgica, los pacientes serán refractarios al tratamiento puramente antipsicóticos

Javitt (1994) y
Heresco-Levy (1996, 1999) han utilizado glicina como potenciador del
tratamiento antipsicótico en pacientes esquizofrénicos; con ello encontraron
una franca mejoría de la sintomatología negativa. Aunque lo anterior plantea la
posibilidad del uso clínico de agonistas NMDA, no debe olvidarse el potencial
citotóxico de estos compuestos.
El glutamato es un
neurotransmisor excitatorio capaz de actuar sobre cualquier neurona cerebral.
Existen cinco vías glutamatérgicas específicas, con relevancia en la
fisiopatogenia de la Esquizofrenia.
Vías corticotroncoencefálicas
Vía descendente que
tiene un papel esencial en la regulación de liberación de neurotransmisores. Se
proyecta desde las neuronas piramidales del cortex prefrontal a centros del
troncoencéfalo:
- Núcleos del rafe: responsables de la neurotransmisión serotoninérgica.
- Locus coeruleus: encargado de la neurotransmisión noradrenérgica
- Sustancia negra: con neurotransmisión dopaminérgica.
- Área tegmental ventral: con neurotransmisión dopaminérgica. Actúa indirectamente en este área a través de interneuronas inhibitorias gabaérgicas, frenando de esta manera la vía dopaminérgica mesolímbica con una inhibición tónica de la liberación de dopamina.
Tras varias observaciones,
se desarrolló una hipótesis en la que los receptores NMDA, específicamente en
las proyecciones corticoencefálicas, podrían ser hipoactivos en la
Esquizofrenia, resultando una hiperactividad dopaminérgica mesolímbica con
aparición de sintomatología positiva (Javitt y Zukin, 1991; Javitt y Coyle,
2004; Pariante et al., 2004).
Asimismo, se
observó que cuando los receptores NMDA son hipofuncionantes debido a la acción
del antagonista fenciclidina (PCP), además de los síntomas positivos descritos aparecían
síntomas negativos, cognitivos y afectivos típicos de la Esquizofrenia.
Esto se debe a que las
neuronas glutamatérgicas córtico-troncoencefálicas actúan como un acelerador de
las neuronas dopaminérgicas mesocorticales, a diferencia de las neuronas
glutamatérgicas córtico-troncoencefálicas sobre las neuronas dopaminérgicas
mesolímbicas, donde actúan por medio de interneuronas gabaérgicas como antes se
ha señalado.
Es por ello, que la
hipofunción del receptor NMDA en las proyecciones córtico-troncoencefálicas
conllevaría una hipoactividad en la vía mesocortical dopaminérgica, explicando
de esta manera la aparición de síntomas negativos, afectivos y cognitivos en la
Esquizofrenia
Vía córtico-estriada
y córtico-accumbens y vías tálamocorticales: Las dos primeras forman parte del brazo
descendente del haz córtico-estriado-tálamocortical (CSTC), mientras que las
terceras constituyen el brazo ascendente de vuelta del mismo.
Habitualmente las
proyecciones glutamatérgicas descendentes finalizan sobre neuronas gabaérgicas en
el estriado, que a su vez proyectan al tálamo creando un filtro sensorial.
La hipofunción del
receptor NMDA en los haces CSTC provoca
la reducción de la función inhibitoria del filtro talámico, lo que puede dar
lugar a un exceso de información sensorial en el córtex, apareciendo de esta
manera síntomas positivos de la Esquizofrenia.
Además de esto, cabe señalar el efecto de la hiperactividad dopaminérgica mesolímbica explicado con anterioridad, que en los haces CSTC reduce aun más la efectividad del filtro talámico, haciendo que demasiada información escape al córtex cerebral de manera difusa, contribuyendo de esta manera a la producción de alucinaciones y otros síntomas corticales como los negativos, afectivos y cognitivos.
 Vías corticotalámicas:
Vías corticotalámicas:
Es una vía
glutamatérgica que aporta entradas sensoriales al tálamo desde el córtex. Una hipofunción
de los receptores de NMDA a este nivel provoca una disregulación de la información
que llega al córtex debido a una sobrecarga y malfuncionamiento de las entradas
glutamatérgicas corticales directamente desde el filtro talámico.
Vías corticocorticales:
Las neuronas piramidales se conectan entre sí mediante glutamato. Teniendo en cuenta la hipótesis de la Esquizofrenia del receptor glutamatérgico hipofuncionante, existiría una comunicación corticocortical disfuncional caótica, pudiendo dar lugar a la aparición de síntomas de Esquizofrenia.
Por último, hacer
mención a la hipótesis excitotóxica de la Esquizofrenia que propone la
neurodegeneración como resultado de una excesiva neurotransmisión excitadora
glutamatérgica, para explicar el curso en declive de esta enfermedad. Esta excitotoxicidad
parece ser la vía final común de muchostrastornos neurodegenerativos
neuropsiquiátricos.
Se cree que la actividad
glutamatérgica excitadora normal se altera, comenzando un proceso patológico de
sobreexcitación que puede acompañarse de diversos síntomas, y que finalmente
conlleva a la muerte neuronal. La excitación neuronal limitada mediada por los receptores
NMDA de glutamato es necesaria para la potenciación a largo plazo, formación de
memoria y sinaptogénesis, siendo útil para llevar a cabo el podado dendrítico
que trata de deshacerse de la materia cerebral muerta (Goff y Wine, 1997;
Konradi y Heckers, 2003).
Podemos concluir que
la hipofunción glutamatérgica podría servir de nexo entre distintos modelos etiopatogénico,
como la teoría dopaminérgica, el neurodesarrollo; la plasticidad sináptica
disfuncional y la hipótesis degenerativa.
Hipótesis Serotoninérgica
Las hipótesis que
implican a la serotonina en la Esquizofrenia, señalan su papel trófico en el
neurodesarrollo, su interacción con el sistema dopaminérgico y los efectos de
la serotonina en la corteza prefrontal a través de sus receptores 5HT2A (Kapur
y Remington, 1996). En los últimos años se ha sugerido un aumento del tono
serotoninérgico central en los pacientes con Esquizofrenia (Abel et al., 1996;
Monteleone et al., 1999).
Diversos autores han
sugerido que la sintomatología negativa de la Esquizofrenia reflejaría en
parte, una hipofunción dopaminérgica en la corteza prefrontal, debida al efecto
inhibidor que tendría la serotonina a ese nivel (Weinberger y Berman, 1988;
Weinberger y Lipska, 1995; Davis et al., 1991).
Es por ello, que los
fármacos inhibidores de la función serotoninérgica desinhibirían la transmisión
dopaminérgica en el córtex prefrontal, mejorando la clínica negativa (Kapur y Remington,
1996; Sepehry et al., 2007).
 Sistema Colinérgico
Sistema Colinérgico
La administración
de agonistas colinérgicos provoca un aumento de sintomatología negativa en los pacientes
con Esquizofrenia y una aparición de estos síntomas en los sanos (Peralta y
Cuesta, 1995), sosteniendo algunos autores, que la hiperactividad colinérgica media
en la sintomatología negativa de la Esquizofrenia (Tandon y Greden, 1989; Davis
et al., 1991).
Sistema Gabaérgico
Estudios postmortem
de pacientes con Esquizofrenia han encontrado niveles reducidos en el cortex prefrontal
de la decarboxilasa del ácido glutámico, que es el indicador de la síntesis de
GABA (Lewis et al., 2005). Además, los receptores de GABA estarían regulados al
alza como mecanismo compensador a los
menores niveles de GABA (Jarskog et al., 2007). Además de los sistemas de
neurotransmisión señalados anteriormente, la Esquizofrenia se ha relacionado con
el ácido gamma-aminobutírico (GABA), neuropéptidos, receptores adrenérgicos y
mensajeros secundarios.



buena info
ResponderBorrar