Narcolepsia
Narcolepsia
Introduccion:
El
término narcolepsie fue introducido por Gelineau en 1880 para englobar los dos
síntomas
clave
del síndrome: hipersomnia y cataplejía. Tradicionalmente, se ha distinguido
entre narcolepsia con cataplejía y narcolepsia sin cataplejía.
La
prevalencia de la narcolepsia típica oscila entre el 25 y 50 por cada 100.000
habitantes.
Beusterien(1999)
Esta patología afecta de manera importante a los pacientes en susrelaciones
personales y laborales, pudiendo ser causa de desempleo, pérdida de autoestima
y otros efectos. SSegún diferentes estudios, la narcolepsia afecta más a la
calidad de vida que la enfermedad de Parkinson o la epilepsia.
Recuento neuroanatomico y neuroquímico
En
1998, dos grupos de investigación independientes identificaron las
hipocretinas.
La
hipocretina 1 y 2, de 33 y 28 aminoácidos respectivamente, son sintetizadas en
un pequeño grupo (10.000 a 20.000 neuronas) bilateral y simétrico de neuronas
polimorfas localizadas exclusivamente en la región dorsal, posterior y lateral
del hipotálamo
Estas
neuronas forman un sistema de proyección difusa que utiliza a las hipocretinas
como neuromoduladores y proyecta a diversas regiones del sistema nervioso
central (SNC)
Por
su localización en el hipotálamo lateral y su homología con la hormona
secretina, se denominó a los péptidos hipocretinas 1 y 2.
El
conocido papel de la región hipotalámica lateral en el control del apetito y el
aumento del consumo de alimento luego de la inyección intraventricular de estos
neuropéptidos, llevó a otro grupo a denominarlos orexinas A y B.
Existen
dos tipos de receptores que son proteínas transmembrana acopladas a proteínas
G. El receptor tipo-1 (rHcrt-1) tiene 10 a 100 veces mayor afinidad por la
Hcrt-1 que por la Hcrt-2, mientras el receptor tipo-2 (rHcrt-2) une ambas con
igual afinidad.
Existe
una distribución diferente de ambos receptores en distintas regiones del SNC,
Las
hipocretinas producen un efecto excitatorio actuando a través del rHcrt-1 en
todos los casos estudiados.
Lo mismo sucede actuando sobre el rHcrt-2, aunque se sugiere también que estos podrían actuar como autorreceptores inhibitorios en las neuronas hipocretinérgicas
Como
consecuencia de los descubrimientos en modelos animales se comenzó a estudiar
el sistema hipocretinérgico en la narcolepsia humana.
El
primer estudio fue realizado por Nishino y colaboradores (2001) en pacientes
caucásicos, observando que los niveles de Hcrt-1 en líquido cefalorraquídeo
(LCR) resultaron fácilmente medibles en muestras control, mientras 90% de los
pacientes con narcolepsia-cataplejia carecía de Hcrt-1 en el LCR (la Hcrt-2 por
su mayor labilidad no se ha logrado medir consistentemente en el LCR).
Estudios
de hibridación in situ e inmunohistoquímica en muestras post-mórtem del
hipotálamo de pacientes con narcolepsia-cataplejia evidenciaron marcada
disminución o ausencia de neuronas hipocretinérgicas.
En
cambio, solo se encontró una mutación en el gen de la hipocretina en un raro
caso narcolepsia-cataplejia de gran severidad, originada a los 6 meses de edad.
Por
lo tanto, aunque existe heterogeneidad en el sector del sistema
hipocretinérgico afectado en los distintos casos de narcolepsia-cataplejia, en
la mayoría de los casos existe una destrucción de las neuronas
hipocretinérgicas, lo que determina una disminución de los niveles de Hcrt-1 en
el LCR.
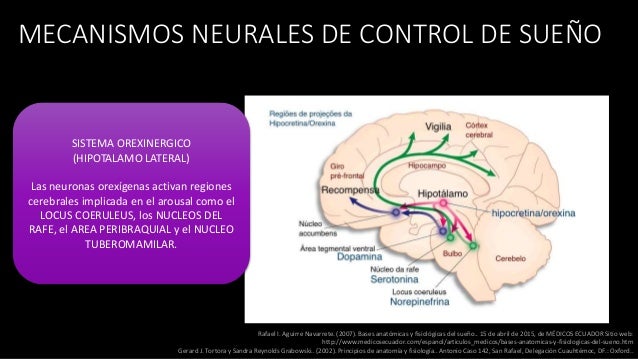
Clasificación de la
narcolepsia:
La
narcolepsia es una enfermedad crónica, relacionada con la alteración de los
mecanismos que regulan el sueño (sistema
hipocretina/orexina).
Según
la segunda edición de la clasificación internacional de los trastornos del
sueño (ICSD) de la American Academy of Sleep Medicine del año 2005 pertenece al
grupo de las hipersomnias de origen central.

Signos y síntomas de la
narcolepsia:
Los
cinco principales síntomas de la narcolepsia son la excesiva somnolencia diurna
(ESD), la cataplejía, la alteración del sueño nocturno, las alucinaciones
hipnagógicas y la parálisis del sueño. Sólo el 10-15% de los pacientes
presentan todos los síntomas.
Excesiva somnolencia diurna (ESD)
Es
un síntoma definitorio de la narcolepsia que está presente en el 100% de los
pacientes y el primero en aparecer10. Se manifiesta como
ataques de sueño irresistibles, principalmente durante las actividades
monótonas y sedentarias. En ocasiones estos ataques se dan en situaciones
inusuales como al caminar, hablar o conducir. Su duración suele ser de 20
minutos o menos. La principal característica, que la diferencia de otras
patologías, es que los ataques de sueño son reparadores permitiéndole reanudar
sus actividades, aunque al poco tiempo reaparece la somnolencia. La ESD
persiste durante toda la vida, diariamente, fluctuando en periodos de semanas o
meses, aunque puede disminuir con la edad.
Cataplejía
Es
el síntoma más específico de la narcolepsia. Se define como atonías musculares
bilaterales (parciales o completas) súbitas, con conservación de la conciencia,
desencadenadas por emociones (risa, alegría, sorpresa, ira, etc.) que revierten
de forma espontánea. La cataplejía parcial se restringe a la pérdida del tono
de un grupo muscular específico como caídas de la cabeza o mioclonías faciales.
Durante
los episodios de cataplejía se observa parálisis flácida, arreflexia y ausencia
de respuesta a la estimulación plantar que fisiopatológicamente corresponden a
la atonía característica del REM pero en vigilia. Los síntomas asociados pueden
ser dificultad respiratoria, sudoración, palpitaciones y ocasionalmente se
superpone una actividad motora fásica a la atonía muscular provocando
fasciculaciones o temblores que pueden confundirse con convulsiones,
permaneciendo el paciente estable sin afectación cardiovascular o respiratoria.
La
cataplejía se da aproximadamente en el 60-90% de los narcolépticos y
puede debutar conjuntamente con la ESD o aparecer entre 1 y 20 años después con
una media de 5-6 años. Su frecuencia es variable y oscila desde un episodio al
mes hasta más de diez episodios al día de diversa intensidad22, con
una duración de unos pocos segundos hasta unos minutos. Aunque excepcionalmente
pueden repetirse durante varias horas o días, lo que se conoce como estado de
mal catapléjico, esta situación se da principalmente en retiradas bruscas de
medicación anticatapléjica.
Parálisis del sueño
Su
frecuencia oscila entre el 25-60% de los pacientes
narcolépticos. Consiste en incapacidad generalizada para moverse o hablar
durante el inicio del sueño o al despertarse, respetando musculatura ocular y
respiratoria, sin pérdida de la conciencia, lo que causa una gran ansiedad. Al
igual que la cataplejía se considera una intrusión parcial del sueño REM en
vigilia. Su duración es de 20 a 30 segundos, es transitoria y cede
espontáneamente con estímulos al paciente. Puede darse en sujetos sanos con o
sin antecedentes familiares de parálisis de sueño o narcolepsia.
Alucinaciones hipnagógicas e hipnopómpicas
Se
producen en el 30-65% de los pacientes . Son fenómenos
pseudoalucinatorios generalmente visuales seguidos de auditivos y cinestésicos
(sensación de que alguien le toca). Usualmente se reconocen como irreales,
presentándose en la transición vigilia-sueño (hipnagógica), al despertar
(hipnopómpica) o en siestas diurnas y no suelen durar más de un minuto.
Episodios de comportamiento automático
Su
prevalencia estimada es alrededor del 40%23. Son acciones
semideliberadas (lenguaje y escritura inapropiado, colocar objetos en lugares
equivocados o la conducción de un vehículo sin conocer exactamente el destino)
con posterior amnesia parcial o total, que se dan en los estados de somnolencia
que preceden a los ataques de sueño. Su duración suele ser de minutos, aunque a
veces duran más de una hora.
Sueño nocturno fragmentado:
Afecta
aproximadamente al 75% de los pacientes , considerándose también uno
de los principales síntomas. En estos pacientes la cantidad total de sueño en
24 horas es igual a la de cualquier persona pero su calidad es distinta. Tienen
un sueño superficial debido a despertares frecuentes breves, sin que el
paciente sea consiente de ello, aunque a veces pueden durar hasta una hora.
Otras patologías asociadas como la apnea del sueño, algunas parasomnias como el
trastorno de conducta del sueño REM (TCR), el sonambulismo y los movimientos
periódicos de las piernas (MPP) contribuyen a la fragmentación del sueño. En
general no tiende a mejorar espontáneamente con el tiempo.
Otros síntomas de la enfermedad
Son
menos específicos, no esenciales para el diagnóstico, entre ellos el trastorno
de conducta REM (TCR), parasomnia que se caracteriza por la pérdida de la
atonía muscular característica del sueño REM, con actividad del sistema motor
de carácter violento y agresivo. En los narcolépticos los episodios de TCR son
menos violentos y su edad de inicio es más baja (25 a 30 años) ayudando a
predecir la narcolepsia en una tercera parte de los pacientes.
Otros síntomas adicionales son la cefalea, las dificultades de memoria y
concentración y la depresión.
Narcolepsia de tipo 1
La narcolepsia de tipo 1 está bien definida desde el punto de vista biológico y patogénico y se relaciona con el déficit de hipocretina, demostrable en el líquido cefalorraquídeo (LCR).
Etiopatogenia
La
narcolepsia de tipo 1 está causada por un trastorno del sistema de la
hipocretina/orexina. Se encontró primero en los animales con narcolepsia
hereditaria la presencia de mutaciones en el gen del receptor de hipocretina 2,
y más tarde en los humanos con narcolepsia adquirida una pérdida selectiva y
grave de las neuronas localizadas en el hipotálamo lateral que producen ese
neurotransmisor.
En
la narcolepsia canina y de otros animales, las neuronas productoras de hipocretina
están preservadas y, por tanto, los niveles de hipocretina en el LCR son normales; el problema radica en los
receptores.
En
la narcolepsia humana, la destrucción de las neuronas del sistema de la
hipocretina hace que su nivel en el LCR sea bajo, pero sus receptores están
conservados. Un fármaco agonista directo de esos receptores podría ser un
tratamiento sintomático eficaz.
La
causa de la neurodegeneración se desconoce. La narcolepsia es, en su mayor
parte, esporádica. Tiene una importante relación con ciertos factores genéticos
que son reguladores de la inmunidad, pero solo se ha descrito un caso con una
mutación en elgen del receptor.
El
98% de los pacientes con narcolepsia tienen el haplotipo HLA-DQB1*06:02, la
asociación entre enfermedad y HLA más fuerte conocida hasta la fecha.
DQB1*06:02 se acompaña casi siempre del alelo DQA1*01:02 en estos pacientes.
Pero
otros factores deben de jugar un papel, ya que este alelo también se encuentra
en el 12-30% de la población general.
Se han descrito, especialmente al inicio de la enfermedad, nivelesbmás elevados que en la población general de anticuerpos contra la proteína TRIB2 (presente en neuronas hipocretinérgicas) y antiestreptocócicas. Esto último contribuye a las hipótesis que sugieren que un estímulo inmune podría precipitar la aparición de la enfermedad.
Esta
enfermedad se expresa típicamente entre los 12 y 25 años y está estrechamente
asociada con el HLA-DR2 y DQB1*0602; la edad de comienzo y la asociación con el
complejo mayor de histocompatibilidad sugiere una etiología autoinmune
|
Estudios en inmunogenetica |
|
Asimismo, se han reportado hallazgos consistentes con un patrón de herencia dominante (con penetrancia incompleta) de un hipotético gen de susceptibilidad para la enfermedad.8 Se han señalado, en particular, tres loci del mapa genético asociados con la narcolepsia: En
21q22.3 — NLC1A, Narcolepsy candidate region gene 1A. En
17q21 — HCRT, OX Hypocretin.10 En
4p13-q21 — NRCLP2 Narcolepsy 2 |
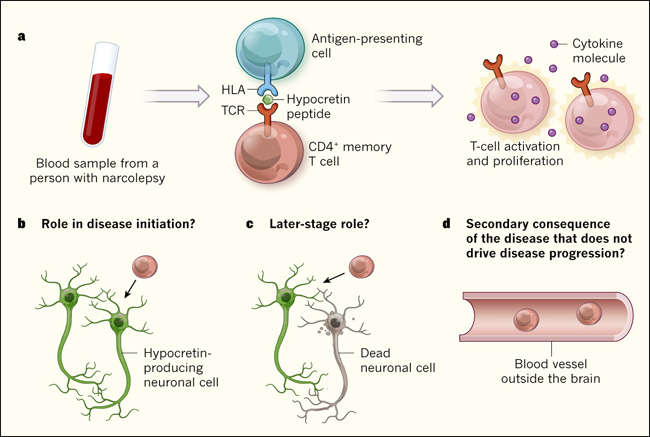
- Fisiopatología
El
defecto básico radica en el sistema de la hipocretina/orexina, que también
tiene relación con la regulación del apetito y el consumo de energía. Hay dos
péptidos (hipocretina 1 y 2) que son producidos por unos pequeños núcleos
situados en el hipotálamo posterior y lateral. Sus proyecciones son muy amplias
a zonas críticas en la regulación de la conducta y el sueño, tales como el
sistema límbico, los sistemas monoaminérgicos (locus coeruleus, núcleos del
rafe, área ventral tegmental, sustancia negra), los núcleos histaminérgicos
tuberomamilares, los núcleos intrahipotalámicos y otros.
La
mayor proyección extrahipotalámica es al locus coeruleus. Hay dos tipos de
receptores acoplados a proteína G: el tipo 1 (Hcrtr 1), que liga exclusivamente
el péptido hipocretina 1, y el tipo 2 (Hcrtr 2), que liga con las dos
hipocretinas.
En
el cerebro de los narcolépticos humanos hay una depleción muy importante de
hipocretina y del ARN mensajero preprohipocretina. El nivel de hipocretina 1 es
muy bajo en el LCR. Este hallazgo se correlaciona muy bien con los otros dos
rasgos básicos de la narcolepsia, que son la cataplejía y el haplotipo DQB1.
También
se relaciona con el acortamiento de la latencia del REM (SOREM). Los pacientes
sin cataplejía y negativos para ese haplotipo tienen niveles de hipocretina 1
en el LCR próximos a los normales.
La
función normal de la hipocretina, gracias a las amplias proyecciones citadas
anteriormente, es contribuir al mantenimiento de la vigilancia durante el día y
a suprimir la aparición del sueño REM. En la narcolepsia se generan las fases
normales del sueño, pero mal reguladas, en particular la fase REM.
La
latencia del sueño REM está acortada, tanto en el sueño nocturno como en el de
las siestas diurnas. Las fases del sueño no están, a veces, bien definidas, y
aparecen períodos de sueño con características intermedias entre el sueño y la
vigilia, como en el síndrome del REM sin atonía, y también se observan
episodios de sueño «ambiguo», con características intermedias entre el REM y el
NREM.
La
crisis de cataplejía corresponde, fisiopatológicamente, a una intensa
inhibición postsináptica de las motoneuronas del asta anterior de la médula,
del mismo tipo que induce la hipotonía fisiológica del sueño REM. Esta
inhibición está mediada por la actividad del núcleo magnocelular bulbar.
Durante la crisis catapléjica, los reflejos musculares, la onda F y el reflejo
H están deprimidos o abolidos.
Neuropatología
Salvo
la pérdida neuronal en los núcleos secretores de hipocretina en el hipotálamo,
no hay otras lesiones neuropatológicas.
Thannickal
y colaboradores(2000) estudiando la anatomía patológica de cerebros de
pacientes con narcolepsia-cataplejia, describieron un aumento de gliosis en el
hipotálamo lateral, consistente con un proceso degenerativo como causa de la
pérdida de las neuronas hipocretinérgicas.
Este
proceso degenerativo es específico para las neuronas hipocretinérgicas ya que
no existe degeneración en otras neuronas de la región. Se sostiene que este
proceso degenerativo sería de causa autoinmune
También
puede ser diagnosticada en la infancia. La primera manifestación suele ser la
hipersomnia, y a continuación, en un plazo de 1 a 2 años, se suma la
cataplejía. El comienzo inverso, primero la cataplejía, se produce en un 10-15%
de los casos, más a menudo en los de inicio infantil.
Muy
raramente, entre el comienzo del primer síntoma y el desarrollo completo del
síndrome pueden pasar hasta más de 20 años. La combinación de la hipersomnia,
la cataplejía y las alucinaciones está presente en más de la mitad de los
pacientes.

Narcolepsia de tipo 2
La narcolepsia de tipo 2 está peor
definida. Su frecuencia es desconocida, pero se estima entre un 10 y un 35% de
todos los casos de narcolepsia. La mayoría de los casos son idiopáticos y
esporádicos, aunque también hay formas familiares o secundarias a una
enfermedad neurológica o una lesión en el SNC.
La presentación clínica es similar a
la de la narcolepsia de tipo 1, a excepción de la cataplejía (por definición). La
fisiopatología se desconoce, aunque es probable que algunos de los casos
diagnosticados como narcolepsia de tipo 2se deban a una privación de sueño no
reconocida.
En cuanto a la etiopatogenia, hay un
estudio que demuestra una pérdida del 33% de las neuronas secretoras de
hipocretina respecto a los sujetos normales (mientras que en la narcolepsia de
tipo 1 sería de al menos el 90%).
Los criterios diagnósticos para la
narcolepsia de tipo 2 son:
- Períodos
diarios de necesidad irresistible de dormir o de quedarse dormido desde al
menos 3 meses.
- Ausencia
de cataplejía.
- La
concentración de hipocretina en el LCR no se ha medido o la concentración
de hipocretina medida en el LCR es de 110 pg/ml o mayor, o mayor de un
tercio del valor medio obtenido en sujetos normales con el mismo ensayo
estandarizado.
- La hipersomnia y/o los hallazgos del TLSM no se explican mejor por otras causas, como el sueño insuficiente, apnea obstructiva del sueño, síndrome del retraso de fase, efectos de la medicación/sustancias o su abstinencia.
Nota Importante:
La
narcolepsia es rara en los niños y tiene algunas características peculiares.
Comienza entre los 6 y los 13 años y suelen tener cataplejía. Asocian
frecuentemente alteraciones del rendimiento escolar, obesidad y síndrome de
alimentación nocturna.
Al
contrario que los adultos, no es raro que tengan dificultades para despertar
por la mañana y, con frecuencia, tienen largos períodos de subvigilancia y
conducta automática. Algunos presentan pubertad precoz.
El
nivel de hipocretina en el LCR suele ser muy bajo, a veces indetectable. Es
importante el diagnóstico y el tratamiento precoces para evitar las
consecuencias negativas para el desarrollo del niño.

Pruebas de diagnostico:
Test de latencia
múltiple de sueño (TLMS)
En
la actualidad es la prueba complementaria básica para el diagnóstico y mide la tendencia al sueño y la aparición o
no de sueño REM.
Consiste
en 5 siestas de 20 minutos cada una con intervalos de 2 horas, previo estudio
polisomnografico, en el que se registran canales de electroencefalografía,
electrooculograma, electromiograma de mentón y electrocardiograma.
Los
pacientes no deben realizar el día anterior actividades de gran esfuerzo y se
deben retirar los medicamentos estimulantes y supresores de REM 2 semanas
antes.
El
resultado del test se considera como patológico si la latencia de sueño es
menor o igual a 8 minutos y la presencia de dos o más entradas en REM25.
En
los narcolépticos la latencia media es de 3,1+/- 2,9 minutos siendo en el 60%
menor de 5. Además un 15% de los pacientes no tienen sueño REM
Polisomnografía (PSG)
La
PSG, además de ser un requisito previo al TLMS para asegurar una buena
eficiencia de sueño, permite descartar otros trastornos del sueño que causen
ESD o que se asocian esta patología, fundamentalmente el síndrome de
apnea-hipoapnea obstructiva del sueño (SAHS), mostrándonos además la fragmentación
del sueño nocturno, la presencia de mayor sueño superficial principalmente fase
1 y la baja eficiencia de sueño con despertares frecuentes.
En la narcolepsia lo específico en la PSG es una latencia de REM menor de 20 minutos en el 40-50% de los pacientes y una latencia de sueño menor de 10 minutos22. Además, es importante evitar el uso de estimulantes o antidepresivos que pueden llevar a falsos positivos tanto en la PSG como en el TLMS.
Solo
en el caso de agravamiento de los síntomas o aparición de otros nuevos debe
contemplarse la realización de otra PSG, que no está indicada en la evaluación
de respuestas a tratamientos en genera

Medidas en líquido cefalorraquídeo (LCR) de Hcrt1
Se
miden los niveles de Hcrt 1 porque además de ser la más específica es la única
que se puede determinar.
Es
una técnica no estandarizada de la que no se dispone en todos los centros. Se
considera diagnóstica cuando es inferior a 110 pg/ml, siendo prácticamente inexistente
en el LCR de pacientes narcolépticos.
Los resultados entre 110 y 200 pg/ml son
inciertos. Es muy específica en pacientes narcolépticos con cataplejía (99%)
pero la sensibilidad es baja (16%) en casos sin cataplejía o con cataplejía
atípica. Además puede estar alterada en otras patologías como el síndrome de
Guillain Barre.No es sensible a fármacos psicotrópicos u otros trastornos del
sueño comunes. Las principales indicaciones son TLMS dudosos y en niños a
quienes no se les puede realizar un TLMS.
También
se puede realizar a pacientes con diagnóstico reciente de narcolepsia con
cataplejía con TLMS negativos, pacientes que no pueden suspender los
tratamientos psicótropos y fallos en el tratamiento.
Tipificación HLA-DQ para DQB 1*0602
Estos alelos son positivos en el 90% de pacientes con narcolepsia con cataplejía, y en el 70% en narcolepsia familiar. Pero en narcolepsia sin cataplejía disminuye al 40% y se da en un 24% de pacientes normales por lo que su especificidad es muy baja, del 8% al 38%. Esta técnica no está incluida en los criterios diagnósticos de narcolepsia
Diagnóstico diferencial
Patologías que cursan
con ESD
-
Hipersomnia idiopática (HI). ESD sin otra causa que lo justifique. Su principal
diferencia con la narcolepsia sin cataplejía es que sus siestas no son
reparadoras. En el TLMS la latencia de sueño es menor de 10 minutos sin ser tan
bajas como en la narcolepsia y no hay entradas en REM. Respecto a la PSG es
normal y existe poca asociación con el HLA. En la HI recientemente se ha
incorporado como síntoma sugestivo en el ICSD-2 la dificultad prolongada en el
despertar por la mañana y un sueño nocturno prolongado mayor de 10 horas.
- Síndrome de apnea-hipoapnea obstructiva del sueño (SAHS). Caracterizado por ESD, trastornos cognitivos, respiratorios, cardiacos, metabólicos o inflamatorios secundarios a episodios repetitivos de obstrucción de la vía aérea superior en el sueño27. En el TLMS puede haber latencias de entrada en sueño inferiores a 5 minutos y 2 o más entradas a REM. Su prevalencia es mayor en narcolépticos. Otros trastornos respiratorios pueden asociar ESD, como el síndrome de hipoventilación alveolar central, síndrome de hipoventilación por obesidad, el asma y la enfermedad pulmonar obstructiva crónica.
-
Trastornos del ritmo circadiano. El trastorno de retraso o de adelanto de la
fase del sueño entre otros descritos en la ICSD-2.
-
Movimiento periódico de las piernas. Esta patología produce frecuentes
despertares que inducen una ESD. Su prevalencia aumenta en los narcolépticos.
-
Síndrome de Kleine-Levin. Es un síndrome autosómico dominante que cursa con
ESD, hiperfagia y conducta sexual desinhibida.
- Alteración del comportamiento
por sueño insuficiente.
-
Distrofia miotónica de Steinert tipo I. Presentan ESD hasta en un 77%, algunos
por SAHS o hipoventilación alveolar crónica. Sin embargo, el hecho de que la
ESD no siempre desaparece al corregir estos trastornos ha sugerido un origen
central por alteraciones del sistema hipotálamo-hipófisis con disminución de la
hipocretina. Estos pacientes presentan alteraciones del REM en TLMS.
-
Insomnio letal familiar. Enfermedad priónica que cursa con alteraciones del
ciclo sueño/vigilia produciendo pérdida progresiva del sueño, periodos breves
de ESD y pérdida del ritmo circadiano, asociándose a alucinaciones complejas, y
entradas súbitas en REM.
-
Somnolencia de causa psiquiátrica. El 10% de los pacientes que consultan por
somnolencia diurna excesiva presentan una patología psiquiátrica,
principalmente psicosis, procesos adaptativos, ansiedad y depresión.
Patologías que cursan
con cataplejía
-
Narcolepsia secundaria. Ataques de sueño y cataplejía con o sin los demás
síntomas de la narcolepsia secundarios a lesiones estructurales en diencéfalo y
tronco encefálico superior y en ocasiones a trauma craneoencefálico. La
presencia de otros síntomas de afectación del SNC indican una narcolepsia
secundaria.
-
Encefalitis paraneolásica anti Ma2. Etiología autoinmune. En ocasiones con
cataplejía y disminución de la hcrt 1 en posible relación a daño hipotalámico.
-
Síndrome de Prader Willi. Enfermedad genética que se caracteriza por obesidad,
retraso mental, talla baja y hipotonía pre y pos natal entre otros, presenta
alteraciones en el funcionamiento del hipotálamo asociándose ocasionalmente a
episodios de cataplejía y disminución de los niveles de hcrt 1.
-
Enfermedad de Norrie (ceguera congénita debida a displasia vitreoretinianal
ligada al cromosoma X que afecta solamente a varones) y síndrome de Moebius
(enfermedad congénita caracterizada por falta de desarrollo del 6o y 7o par
craneal) se asocian a daño a nivel cerebral medio y hipotalámico presentando en
ocasiones episodios de cataplejía like y trastornos del sueño.
-
Enfermedad de Nieman Pick tipo C. Enfermedad de depósito graso por un trastorno
genético del metabolismo, que cursa con cataplejía y disminución de los niveles
de Hcrt 1.
-
Epilepsia. Ocasionalmente convulsiones subclínicas y la ESD por medicación de
pacientes epilépticos pueden confundirse con narcolepsias o viceversa,
episodios de parálisis del sueño o parasomnias pueden confundirse con
epilepsias.
-
Otros. La cataplejía también debe diferenciarse de crisis atónicas, isquemia
vertebrobasilar en forma de pérdida súbita del equilibrio (dropattacks),
síncopes, lipotimias y parálisis hiper o hipopotasémicas.
Tratamiento Farmacologico:
Hasta hace poco los diferentes tratamientos se
dirigían a controlar por separado cada uno de los síntomas. Recientemente las
nuevas directrices europeas y de la Asociación Americana de Medicina de
Sueño (AASS) incorporan nuevos fármacos capaces de controlar los
principales síntomas de la narcolepsia.
Mientras
que para unos la fragmentación del sueño nocturno es una molestia grave, para
otros lo es la cataplejía, y para la mayoría la somnolencia.
El
oxibato sódico (3-9 g fraccionado en dos dosis, una al irse a la cama y otra en
la madrugada/media noche) es muy efectivo para tratar la cataplejía y la
alteración del sueño nocturno, y algo menos para la hipersomnia diurna.
El
pitolisant, un agonista/antagonista inverso de los receptores H3 de la
histamina que es efectivo para la somnolencia (al menos igual que modafinilo) y
la cataplejía, ha recibido la aprobación de la Agencia Europea de Medicamentos.
La
dosis inicial es de 9 mg por la mañana hasta un máximo de 36 mg (18 en casos de
insuficiencia renal o hepática). Los efectos adversos leves (insomnio, cefalea,
ansiedad, irritabilidad,etc.) son relativamente frecuentes, pero los efectos
graves son raros.
La
eficacia a largo plazo y el papel definitivo del pitolisant y otros fármacos
del mismo grupo está por determinar con la experiencia.
Otros
fármacos para combatir la hipersomnia son el modafinilo (de 200 a 400 mg/día) o
el metilfenidato (de 10 a 60 mg/ día). El modafinilo es un inductor enzimático
y disminuye el efecto de los anticonceptivos. El uso de D-anfetamina (de 5 a 15
mg/día) conlleva el riesgo de adicción y efectos secundarios cardiovasculares
(HTA, prolongación del QT).
Hasta
la introducción del oxibato, los únicos fármacos eficaces para las crisis de
cataplejía eran los antidepresivos tricíclicos, de los cuales la clomipramina
(de 10 a 100 mg/día) es el primero que debe emplearse, seguido de imipramina o
amitriptilina.
También
suelen mejorar las alucinaciones hipnagógicas y las parálisis del despertar.
Los ISRS (fluoxetina, reboxetina) han sido eficaces en algunos pacientes.
Si
no hay apneas de sueño asociadas y no se usa oxibato, se puede prescribir un
hipnótico no benzodiazepínico o una benzodiazepina de acción muy corta para
mejorar el sueño nocturno.
Si
el paciente tiene otros fenómenos anormales durante el sueño nocturno, como
conducta anormal durante el REM o movimientos periódicos, también se puede
beneficiar de otros tratamientos sintomáticos, como clonazepam o agonistas
dopaminérgicos.

Futuro del tratamiento
de la narcolepsia
Los
siguientes pasos son la búsqueda de fármacos que controlen todos los síntomas
principales de la narcolepsia con eficacia demostrada a largo plazo. Existen
tres tratamientos emergentes principales:
1.
Tratamientos sintomáticos de modulación endocrina/transmisora. Agonistas
selectivos de la histamina (antagonistas H3), antagonistas de la hormona
liberadora del crecimiento (GHRH), agonistas del gamma-hidroxibutirato (GHB) y
agonistas del ácido gamma-aminobutírico tipo B (GABA-B).
2.
Tratamientos basados en la hipocretina. Agonistas de la hipocretina y
trasplantes celulares, orientados hacia la reposición de hipocretina o incluso
prevenir la pérdida de neuronas que contienen este neuropéptido.
3. Tratamientos de base inmunitaria. Corticoides, Ig IV y la plasmaféresis
Referencias bibliográficas:
Beusterien
KM, Rogers AE, Walsleben JA, Emsellem HA, Reblando JA, Wang L et al. Health-related
quality of life effects of modafinil for treatment of narcolepsy. Sleep 1999; 22: 757-765
De
Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, et al. The
hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc
Natl Acad Sci USA 1998; 95(1): 322-7.
De
Lecea L, Sutcliffe JG. The hypocretins/orexins: novel hypothalamic
neuropeptides involved in different physiological systems. Cell Mol Life Sci
1999; 56(5-6): 473-80.
Hungs M,
Mignot E. Hypocretin/orexin, sleep and narcolepsy. Bioessays 2001; 23(5): 397-408.
Kilduff TS,
Peyron C. The hypocretin/orexin ligand-receptor system: implications for sleep
and sleep disorders. Trends Neurosci 2000; 23(8): 359-65.
Lin L, Hungs M, Mignot E. Narcolepsy
and the HLA region. J Neuroimmunol 2001; 117(1-2): 9-20.
Martin G,
Fabre V, Siggins GR, de Lecea L. Interaction of the hypocretins with neurotransmitter
in the nucleus accumbens. Regul Pept 2002; 104(1-3): 111-7.
Mignot E. A
commentary on the neurobiology of the hypocretin/orexin system.
Neuropsychopharmacology 2001; 25(5 Suppl): S5-S13.
Overeem S,
Mignot E, Van Dijk JG, Lammers GJ. Narcolepsy: clinical features new
pathophysiologic insights, and future perpectives. J Clin Neurophysiol 2001;
18: 78-105.
Peyron C,
Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, et al. Neurons
containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci
1998; 18: 9996-10015.
Thannickal TC,
Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, et al. Reduced number
of hypocretin neurons in human narcolepsy. Neuron 2000; 27: 469-74.
Willie JT,
Chemelli RM, Sinton CM, Yanagisawa M. To eat or to sleep? Orexin in the
regulation of feeding and wakefulness. Annu Rev Neurosci 2001; 24: 429-58.
Importancia de las hipocretinas en la patogenia de la narcolepsia http://www.scielo.edu.uy/scielo.php?script=sci_arttext&pid=S1688-03902003000100004



Comentarios
Publicar un comentario