Enfermedad de Huntington
Enfermedad de Huntington
La enfermedad de Huntington (EH) es el prototipo de las enfermedades que
cursan con corea de causa hereditaria. Otras causas de corea hereditaria o
esporádica se recogen en el cuadro
Es relativamente frecuente en la raza caucásica (5-7 casos por cada 100.000 habitantes) y mucho menos en Asia o África.
Hay algunos focos de alta prevalencia, como a orillas del lago Maracaibo, que
fueron determinantes para el descubrimiento del gen de la enfermedad.
Etiología y patogenia
Tiene una herencia autosómica dominante por una repetición anormal del trinucleótido CAG en el exón 1 del gen
de la huntingtina (HTT-cromosoma 4), que se traduce en tractos proteicos
anormalmente largos.
No se conoce completamente la función de la huntingtina, pero se sabe que
tiene implicación en el transporte vesicular y en la red de microtúbulos.

· El
número normal de repeticiones es de 26 (incluido)
· De 27 a 35 hay un rango gris o intermedio en el que el paciente no va a presentar la enfermedad, pero dada la inestabilidad del alelo mutado puede expandirse en las siguientes generaciones.
De 36 (incluido) en adelante se considera que la persona presenta predisposición genética a la EH. Entre 36 y 39, la penetrancia es incompleta, por lo que junto a los alelos intermedios se podrían justificar aparentes mutaciones de novo.
· El
número de repeticiones se relaciona con la edad de inicio de la enfermedad,
aunque no es el único factor determinante.
· Las
formas de comienzo infantojuvenil ocurren a partir de 50-55 repeticiones.
El alelo mutado es un gen inestable que durante los procesos cromosómicos
puede disminuir o aumentar.
Es más frecuente que aumente si el progenitor es varón, porque en la
espermatogénesis se emplea el doble de pasos que en la generación del óvulo.
El fenómeno del mosaicismo explica que una persona con una mutación en el
rango gris en el ADN de la sangre pueda tener células gonadales con un mayor
número de repeticiones, lo que se debe tener en cuenta en el consejo genético.
El aumento del número de repeticiones de generación en generación hace que
se produzca en ocasiones una anticipación, por lo que en los hijos o los nietos
la enfermedad comienza más precozmente que el progenitor inicial.
No se conoce con exactitud el papel de la poliglutamina anormal en la
patogenia de la enfermedad, aunque es muy probable que tenga un efecto
neurotóxico por un mecanismo de ganancia de función. La neurotoxicidad va
ligada a mecanismos excitotóxicos, de estrés oxidativo y de apoptosis.

En animales transgénicos y en el modelo experimental por el ácido nitropropiónico, que tiene un efecto neurotóxico selectivo sobre el estriado, se están investigando los mecanismos de la neurotoxicidad y se están ensayando fármacos o agentes neurotróficos, como el factor neurotrófico derivado del cerebro (BDNF), que la reduzcan.
Estos productos podrían actuar a dos niveles: el primero, impidiendo la
agregación de las proteínas y la formación de los cuerpos de inclusión, y el
segundo reduciendo el efecto neurotóxico de los agregados proteicos anormales.
Otras terapias van encaminadas a la reducción de niveles de huntingtina
para compensar dicha ganancia funcional, como por ejemplo oligonucleótidos sin
sentido que se unen al ARN mensajero e impiden la replicación proteica
(silenciamiento del gen).
Se conocen varias fenocopias (HDL, Huntington’s disease like).
La HDL-4 es la más frecuente. Es alélica de la ataxia espinocerebelosa 17
(SCA17) y se debe a una mutación en el gen de la proteína de unión a TATA. La
HDL-1 es una enfermedad por priones; la HDL-2 tiene una mutación en el gen de
la junctofilina, y en el caso de la HDL-3 se desconoce la base genética.
Las expansiones de un hexanucleótido en C9ORF72 son la causa más frecuente
del síndrome de demencia frontotemporal con esclerosis lateral amiotrófica ,
pero se han descrito recientemente casos que se presentan con corea, distonía,
temblor y rigidez como en una EH.
También imitan a la EH las mutaciones en los genes de las calcificaciones idiopáticas cerebrales, en el gen de la ferritina y en el de la neuroacantocitosis

- Anatomía patológica y neuroquímica
Las principales lesiones se encuentran en la parte anterior del núcleo
caudado y el putamen, y se extienden con un gradiente dorsoventral.
En fases iniciales, la degeneración es muy selectiva de las neuronas
espinosas pequeñas y medianas que son gabaérgicas, contienen encefalina y se
proyectan al globo pálido externo, mientras que se preservan mejor las otras
poblaciones neuronales ricas en neuropéptido Y y somatostatina, que se proyectan
al globo pálido interno.
El resultado de estos trastornos neuroquímicos es doble; por un lado, una
activación de los receptores glutamatérgicos NMDA, lo que puede ser una base
patogénica de la enfermedad y, por otro, la alteración preferente de la vía
indirecta eferente dopaminérgica de los ganglios de la base, lo que se correlacionaría
con la corea como primer síntoma y con la eficacia de los fármacos
antidopaminérgicos para su control.
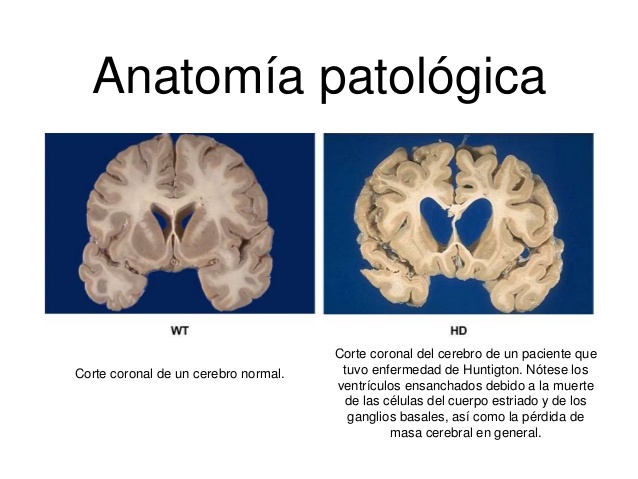
En fases avanzadas, la atrofia del caudado-putamen suele ser tan grande que
se aprecia macroscópicamente por la dilatación del asta frontal de los
ventrículos. También existe una atrofia cortical, sobre todo frontotemporal
anterior, en los casos avanzados.
En la histología se aprecian una pérdida neuronal y gliosis. En las
neuronas se encuentran inclusiones intranucleares inmunorreactivas para
ubicuitina y poliglutamina.
Las inclusiones representan, probablemente, intentos de secuestrar las proteínas
patógenas formando grandes conglomerados inocuos para la neurona. De hecho, en
las zonas histológicamente más anormales suele haber menos inclusiones que en
las mejor respetadas. Las inclusiones están presentes en los cerebros de
individuos presintomáticos fallecidos por otro motivo.
Suele haber lesiones en otras estructuras del tronco cerebral, el tálamo,
el cerebelo y la SN (que pueden explicar en parte el síndrome
rígido-acinético).

Clínica
Los portadores de expansiones en la zona gris deben seguirse regularmente
para detectar la posible aparición de síntomas.
La edad de comienzo de los casos con expansiones en el rango patológico
suele ser la cuarta o quinta década, aunque puede aparecer a cualquier edad. Es
muy rara por debajo de los 2 años y por encima de los 70.
El cuadro clínico se caracteriza por movimientos anormales coreicos o
distónicos, faciales y de las extremidades (59% de los casos), a los que se
añaden trastornos de la memoria y del comportamiento (que son síntomas de
comienzo en el 29% de los casos) y, más tarde, demencia de tipo subcortical o
frontal.
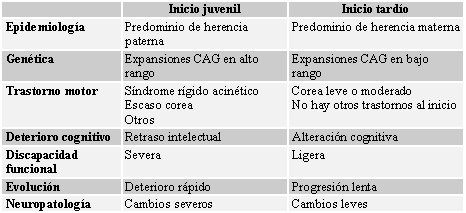
El 10-15% de los casos son de comienzo infantojuvenil, y en ellos aparece
con más frecuencia un síndrome rígido-acinético sin corea. A veces se presenta
con crisis convulsivas remedando una epilepsia mioclónica progresiva
(epilepsia, ataxia y demencia).
Otros datos de presentación de la variedad infantil son el deterioro del
rendimiento escolar, las alteraciones de la marcha y la disfunción oral. El diagnóstico
clínico en estos casos es difícil si no hay una historia familiar que indique
una EH.
La variabilidad clínica de la EH es muy amplia. Los pacientes presentan
alteraciones sutiles motoras o cognitivas en la fase que se denomina «presintomática»
y que sería más correcto llamar «prediagnóstico clínico».
Las que se consideran más precoces son:
a) La alteración de los movimientos oculares sacádicos y el seguimiento
ocular irregular.
b) La adiadococinesia (alteración de los movimientos alternantes
dorso-palma de una mano sobre la otra)
c) Los reflejos musculares vivos con el fenómeno de hung up (la contracción sostenida del cuádriceps al explorar el reflejo rotuliano mantiene la pierna «colgada» o elevada),
d)a irritabilidad y un peor resultado en pruebas como
el test de Stroop son los primeros defectos en estudios neuropsicológicos.
Todos estos hallazgos casi nunca ocurren antes de los 20 años en las formas clínicas habituales, que comienzan a presentar movimientos anormales obvios alrededor de los 30 años.
En la fase de la enfermedad desarrollada, los pacientes muestran trastornos
motores, cognitivos y psiquiátricos:
- Trastornos motores.
Tienen movimientos coreicos o distónicos intensos de la cara, cuello,
tronco y extremidades. Los movimientos coreicos se aprecian durante la marcha en
las extremidades, en la cara, en el cuello y el tronco. Las sacudidas del
tronco y de la pelvis dan a la marcha del paciente un aire grotesco.
La corea aumenta con la distracción. La impersistencia motora los
incapacita para mantener la lengua sostenidamente fuera de la boca, para
apretar la mano del observador sin relajar los dedos o para mantener la mirada
fija en un punto sin dirigirla a cualquier estímulo que aparezca en el campo
visual.
La impersistencia motora de la mano explica que los pacientes dejen caer
las cosas y que al caminar las piernas se les doblen. Los pacientes son torpes
para los movimientos finos con los dedos de las manos.
- Trastornos cognitivos
Al principio no tienen fallos obvios de la memoria episódica o a largo
plazo, pero sí de la atención, concentración, memoria de trabajo y funciones ejecutivas
(planificación, verificación, etc.). Más adelante, el deterioro mental se hace
evidente y aparecen fallos de memoria, del juicio, de las funciones
visuoespaciales y comportamientos obsesivo-compulsivos, hasta la demencia
global.
- Trastornos psiquiátricos
Las tasas de alcoholismo y de abandono del trabajo, altercados sociales y
suicidios son altas. Hasta el 25% de los pacientes intentan el suicidio en algún
momento de la evolución, incluso en fases precoces.
La depresión, el divorcio, la pérdida del trabajo o el aislamiento
incrementan el riesgo de suicidio. Algunos pacientes pueden tener rasgos
psicóticos esquizoides o paranoides y se les diagnostica de esquizofrenia, pero
las alucinaciones son raras.
Algunos casos de comienzo tardío tienen un cuadro moderado tanto en lo que
se refiere a la corea como al deterioro mental. En ocasiones, la corea está
ausente, pasa inadvertida o se considera «senil», por lo que al paciente se le
diagnostica de otro tipo de demencia; esto puede dificultar el diagnóstico correcto
en sus hijos.
Con los años, los movimientos coreicos suelen disminuir de amplitud, la marcha se hace rígida de tipo parkinsoniano, empeoran las posturas distónicas, el lenguaje se hace ininteligible, aparece disfagia y la demencia es profunda, con mutismo e incontinencia. El pronóstico es fatal en unos 15-20 años.

- Diagnóstico y diagnóstico diferencial
El diagnóstico es sencillo cuando la persona que consulta por la corea aporta el antecedente familiar, pero la historia familiar no siempre es positiva, por diversos motivos. El índice de abandonos del hogar por parte del progenitor enfermo es más elevado de lo normal, lo mismo que el de entregas del niño recién nacido en riesgo a una institución.
No es raro que la historia del
progenitor enfermo se limite a que sufrió algún trastorno neuropsiquiátrico
vago o a que murió en un asilo, pero estos datos deben ser suficientes para
sospechar el diagnóstico.
En la TC o en la RM se observan la atrofia de la cabeza de los núcleos
caudados y la dilatación de las astas y surcos frontales. El caudado comienza a
atrofiarse cuando el paciente todavía está asintomático (hasta 15 años antes),
y ya está atrófico en los portadores de la mutación a la edad esperada de
comienzo de la enfermedad (por el tamaño de la expansión).
Un posible marcador biológico de la enfermedad es el nivel de NLP (neurofilament
light protein) en sangre, que está elevado ya en la fase presintomática y se
relaciona con el número de repeticiones. Otras técnicas de RM como la
espectroscopia, la tensión de difusión o la funcional detectan anomalías en la
fase presintomática de la enfermedad.
En la SPECT se demuestra una hipoperfusión frontal. La PET permite observar, además, el hipometabolismo del núcleo caudado incluso en personas presintomáticas.
Este hallazgo no es, sin embargo, específico, y se observa
hipometabolismo del núcleo caudado en otras coreas.

El diagnóstico se confirma por el estudio genético, que también permite
identificar a los portadores presintomáticos de la mutación. Este diagnóstico
anticipado es motivo de controversia, pues se trata de sentar una sentencia en
una persona joven a la que no se le ofrece ninguna posibilidad preventiva ni
terapéutica de un proceso fatal por lo que deben
Recibir un adecuado consejo genético y psicológico por parte de un equipo
multidisciplinario. Solo un 5% de los individuos en riesgo solicitan el
diagnóstico anticipado. La mayor frecuencia se da entre quienes quieren
planificar su descendencia.
A los portadores se les puede ofrecer un embarazo con óvulos o espermatozoides donados y, en algunos centros, un diagnóstico preimplantacional. Los programas de consejo genético y diagnóstico anticipado en la EH han demostrado ser rentables y beneficiosos para los que reciben un resultado positivo.

- Tratamiento
No hay tratamiento específico para la EH. La corea mejora con antidopaminérgicos, de los que está específicamente indicada la tetrabenazina (empezando por 25 mg/día) cuando la corea interfiere en la marcha o la deglución o es muy intensa.
Pero la mejoría de la corea no siempre se acompaña de una mejo ría funcional. La tetrabenazina induce depresión, que debe ser vigilada por el riesgo de suicidio. La tetrabenazina no es antipsicótica, por lo que si la corea se complica con psicosis hay que usar otros fármacos como olanzapina, aripiprazol o risperidona. Si el SP es relevante, se puede utilizar L-DOPA. También puede ser útil la amantadina.
Las BDZ y los antidepresivos pueden ayudar como tratamientos sintomáticos. El riesgo de suicidio, especialmente en el inicio de la sintomatología o en el momento del diagnóstico genético, es elevado y requiere medidas específicas de prevención y tratamiento. Los ISRS y algunos antiepilépticos, como el valproato o la carbamazepina se han propuesto para mejorar la irritabilidad y las ideas obsesivas. El soporte psicológico y la terapia familiar, la rehabilitación física y cognitiva, el seguimiento nutricional, etc. son ayudas esenciales. Muchos enfermos requieren, con el paso del tiempo, el ingreso en una institución especializada.




Comentarios
Publicar un comentario