Etiología de la enfermedad de alzheimer
Etiología de la Enfermedad de alzheimer
La EA es la cuarta causa de muerte en
países desarrollados y la primera causa de demencia, presente entre 40 y 50% de
los casos. Es la más frecuente de las neurodegeneraciones. Hoy en el mundo
existen más de 24 millones de personas con demencia y se estiman 4.6 millones
de casos nuevos cada año.
La presencia de placas seniles (PS) y
ovillos neurofibrilares (ONF) constituyen los marcadores biológicos de la EA. Sin
embargo, la etiología de las formas esporádicas, los mecanismos de acumulación
de las PS y los ONF, la relación entre éstos y su peso en el deterioro cognitivo
permanecen en estudio.
ANATOMÍA PATOLÓGICA
La EA se caracteriza a simple vista
por una afectación cortical con respeto de estructuras subcorticales. Se observa
una disminución de la transparencia y fibrosis de las leptomeninges, con
grandes lagunas subaracnoideas por los espacios dejados entre los surcos
cerebrales.
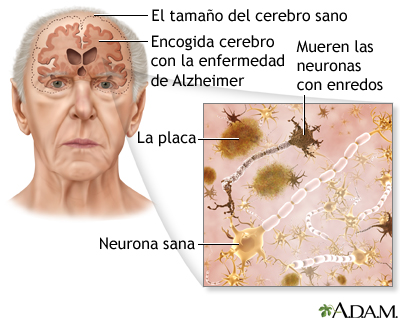
Al retirar las meninges se muestra un cerebro pálido con disminución del peso (aproximadamente 800 g contra 1,300 a 1,700 g en el adulto normal) con atrofia global, bilateral y simétrica de ambos hemisferios, con circunvoluciones atrofiadas y surcos aumentados que en ocasiones dejan ver la profundidad de los valles, con mayor afectación fronto temporo parietal (áreas de asociación) y con respeto relativo de áreas sensoriomotoras primarias y lóbulo paracentral. La región más comprometida es la cara mesial del lóbulo temporal que muestra signos de esclerosis. Se observa un aumento del volumen de los ventrículos.

MICROSCÓPICO
Corroboran los hallazgos antes descritos. Se observan además cambios subcorticales de importancia tales como la despoblación neuronal del núcleo basal de Meynert, los núcleos del rafe, el núcleo ceruleus y lesiones en sustancia blanca.
Las dos lesiones típicas que definen la EA son. Este núcleo se encuentra rodeado por neuritas degeneradas, microglias activadas y astrocitos que le dan un aspecto de nido. Otras sustancias que conforman las PS son la alfa sinucleína (principal componente no amiloide), alfa 1 antiquimotripsina, alfa 2 macroglobulina, la apolipoproteína E, ubiquitina y las presenilinas.

También distinguen neuronas con degeneración
neurofibrilar alrededorpero no en contacto con las placas. Por su aspectose clasifican en:
1. Difusas. Formadas por una delicada
red de finas fibrillas de filamentos de amiloide, sin neuritas degeneradas. Su
centro y sus límites no están bien definidos.
2. Primitivas. Son las más frecuentes.
Se caracterizan por depósitos extracelulares desordenados de Aβ no fibrilar o
escasamente fibrilar. No presentan centro definido pero sus límites son más precisos.
3. Clásicas. También llamadas placas
neuríticas, presentan un centro amiloide y una corona en la periferia compuesta
por astrocitos reactivos, microglía y neuritas distróficas que corresponden a
dendritas y axones degenerados.
4. Quemadas. Sólo presentan un centro condensado de amiloide. No tiene componentes celulares.
Estas formas representan los
diferentes estados evolutivos de las placas, que comienzan con la acumulación difusa
de amiloide, luego éste se organiza y define, asociándose la respuesta inmunológica
y finalmente desaparecen los elementos celulares.
Las PS se encuentran en los cerebros
de personas sin déficit cognitivos, pero en menor proporción.
Concentraciones mayores son criterios
para el diagnóstico patológico de EA. Sin embargo, estudios recientes con
Tomografía por Emisión de Positrones usando PIB para el seguimiento de la
acumulación de PS en vivo demuestran que el aumento en su número ocurre sólo en
los primeros años de evolución. Luego existe una estabilización a pesar de que
el deterioro cognitivo continúa.
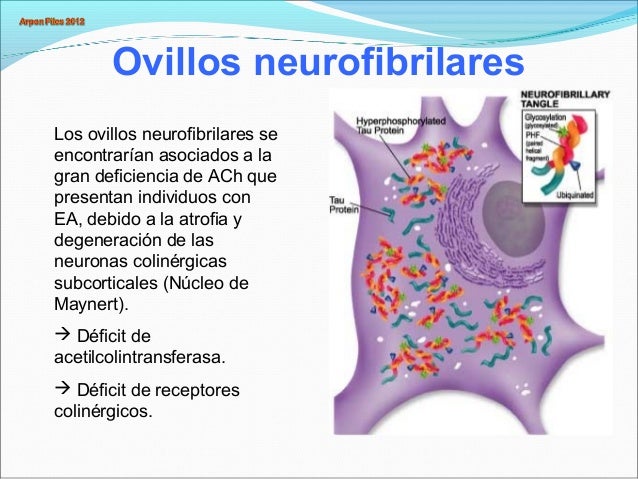
Ovillos Neuro fibrilares (ONF)
Las neuronas presentan acumulación de
inclusiones en forma de llama alargada y a veces forman una cesta alrededor del
núcleo. Son basófilos a la tinción hematoxilina y eosina (HE) y tiñen
fuertemente con tinciones de plata.
Sucesivamente las inclusiones llenan el citoplasma, particularmente en el soma y la dendrita apical facilitando la neuorodegeneración y muerte neuronal principalmente por mecanismos apoptóticos, quedando luego sólo el citoesqueleto (nódulos sepulcrales o fantasma).
El desarrollo de nuevas técnicas de tinción permite visualizar las proteínas Tau fosforiladas cuando aún son solubles, en estados cada vez más tempranos llamados pre-fibrilares, que son la base de la clasificación según los estados evolutivos de Braak, lo que ha sugerido cambios en los criterios diagnósticos patológicos.
Las PS y los ONF no tienen exactamente
la misma distribución ni correlacionan igual con la clínica. Ambas lesiones se
encuentran bien distribuidas en regiones fronto temporales y respetan las áreas
sensoriomotoras primarias; sin embargo, las PS también se encuentran en el neocortex
occipital donde se hallan muy pocos ONF.
Al contrario, los ONF tienen su máxima
concentración en cortex límbico donde se observan pocas PS:
1. Las PS son relativamente poco
frecuentes en estructuras límbicas y más visibles en neocortex.
Correlacionan mejor con la pérdida
sináptica que precede al depósito amiloide y ONF. Por lo tanto, se deduce que
la pérdida sináptica es primaria y no secundaria a la despoblación neuronal. El
BA es neurotrófico en bajas concentraciones y neurotóxico en altas.
2. Los ONF correlacionan mejor con la
despoblación neuronal, el patrón de atrofia y el déficit cognitivo. Se distribuyen
por regiones muy características: Alocortex (entorrinal y perirrinal), región
CA1 del hipocampo y amígdala. También se encuentran en el núcleo basal de
Meynert, Isocortex temporal (áreas 20 y 21 de Brodmann) y el resto de las
estructuras hipocámpicas (CA3, CA4, giro dentado y presubículo).

Se encuentran cada vez con menos
frecuencia en estructuras no límbicas ( Neocortex).
Otros hallazgos menos específicos son:
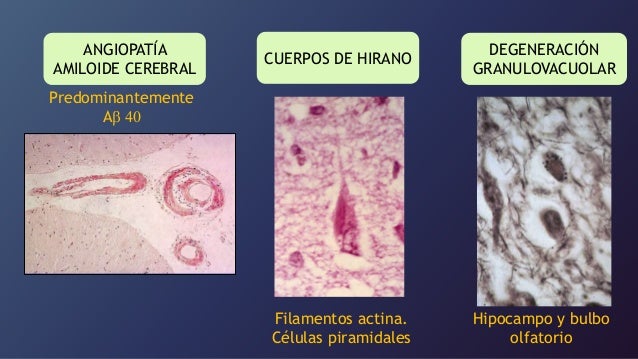
• Degeneración granulovacuolar:
Presencia de vacuolas intra neuronales de 3 a 5 micrones que pueden asociarse o
no a los ONF. Se encuentra mayormente en el hipocampo.
• Despoblación neuronal: Asociada a
los ONF.
• Inclusiones y pigmentos: Lipofucina,
cuerpos de Hirano (actina) y Cuerpos de Lewy (alfa sinucleína).
• Angiopatía congófila: Depósito de
sustancia amiloide alrededor de vasos cerebrales medianos (por fuera de la capa
elástica) y leptomeninges.
• Otros: Satelitosis, neruronofagia y fragmentación son etapas de la muerte neuronal mediada por glías. Depósito de metales en especial el aluminio
Etiopatogenia
La atención se ha centrado en las lesiones típicas y en sus componentes primarios: el péptido BA de las PS (hipótesis de cascada amiloide) y la proteína tau de los ONF (hipótesis de fosforilación Tau).
Metabolismo beta amiloide.
El péptido BA constituye un pequeño
fragmento de una proteína transmembranal de función desconocida (posiblemente participe
en la transducción de señales) llamada proteína precursora amiloide (APP) que
se sintetiza en el cromosoma 21.
Dicha proteína se encuentra en las membranas
citoplasmáticas, endosomal y del sistema de Golgi tanto del sistema nervioso
como de las células sanguíneas. Ciertas isoformas del APP tienen un dominio
inhibidor de la proteasa de Kunitz reguladora de la cascada de la coagulación.
En los sujetos normales el péptido BA
es fragmentado por una proteína secretasa alfa que la divide en dos segmentos
formando la nexina II con acción moduladora de la coagulación y el péptido BA
de 16 aminoácidos altamente soluble.
Este péptido BA se une a la alfa 2
macroglobulina que señaliza a las proteínas que serán degradadas formando un complejo
BA-A2M al que se une una proteasa. El producto de estas interacciones es reintroducido
a la célula nerviosa primero adhiriéndose a éstas a través de la A2M por el
receptor de membrana que es común a la LDL y a la APOE.
Existe una vía alternativa que
consiste en el desprendimiento completo del péptido BA de 40 a 42 aminoácidos.
Esto ocurre por la acción de las beta (escisión 1) y las gamma secretasas
(escisión 40-42).
La amiloidogénesis, que corresponde a
la formación de filamentos de péptido β-amiloide (Aβ) en forma de depósitos
extracelulares a partir de la proteína APP (amyloid protein precursor =
proteína precursora del amiloide). Es
esencialmente el resultado de una disfunción del APP que induce la
neurotoxicidad de Aβ generando depósitos de fibras amiloides insolubles que
provocan la degeneración neurofibrilar.
El APP se libera al espacio extracelular
a través de una secretasa (α). Éste APP no es amiloidogénico.
El APP puede tomar una ruta
alternativa siendo procesado por otro tipo de secretasas (β y γ) produciendo Aβ
que puede permanecer en estado soluble pasando al líquido cefalorraquideo (LCR)
o agregarse formando fibras amiloides insolubles.

Las gamma secretasas están compuesta
por cuatro segmentos: presenilina, nicastrina,
PEN-2 y APH-1, siendo la presenilina su sitio activo.
La acumulación de fragmentos BA 1-42,
insoluble en el intersticio sufre varias transformaciones.
La primera es la pérdida de la
conformación helicoidal (alfa hélice) para pasar a la unión de varios péptidos
(el centro del núcleo está formado por AB 42 al que luego se adiciona el AB 40)
en conformación de hoja plegada, de difícil degradación.
Las mutaciones fuera del segmento que codifica el fragmento Aβ se segregan con otros fenotipos de amiloidosis diferentes de la EA, con angiopatía amiloide y hemorragias cerebrales hereditarias.
Las presenilinas 1 y 2 tienen varias
acciones, entre ellas modular la γ-secretasa (o incluso su misma función), y
esta es la base patogénica del incremento de la amiloidogénesis en los
pacientes portadores de mutaciones en los genes PS1 y PS2.La presencia de estos
cuerpos provocan la activación del sistema inmune, en especial de la
microglias, que perpetúan la lesión por seudo inflamación y liberación de
radicales libres.
Disfunción de τ y degeneración neurofibrilar
La DNF que se observa al microscopio
en el interior de las neuronas está formada por agregados de proteína τ
fosforilada anormalmente.
Existen unas variedades de demencias en las que la formación de la DNF es el acontecimiento patogénico primario, por mutaciones en el gen que codifica para la proteína τ (gen MAPT en el cromosoma 17) o por otras causas, y que ocurre sin depósito de amiloide.
Pero en la EA el acuerdo general es
que la DNF es un proceso patológico secundario al efecto neurotóxico del
depósito de Aβ , aunque se desconoce su mecanismo exacto.
Por los experimentos realizados en
animales transgénicos se sabe que la disfunción de la proteína τ, cuyas
principales funciones son estabilizar los microtúbulos de las prolongaciones
neuronales y facilitar el transporte axoplásmico, comienza muy precozmente,
mucho antes de que se agreguen y formen los filamentos anormales de la DNF.
Las proteínas tau en condiciones normales
estabilizan los microtúbulos. Cuando se encuentran hiperfosforiladas como es el
caso de las que se encuentran en los ovillos neurofibrilares, disminuye su
interacción con los microtúbulos y los desestabilizan.
 Metabolismo neurofibrilar
Metabolismo neurofibrilar
Los ONF están compuestos principalmente
por filamentos helicoidales pareados formados por proteínas Tau
hiperfosforiladas.
También están formados por otras
proteínas como la MAP2 (predomina en dendritas), la ubiquitina, y los péptidos BA
(lo que apoya la teoría de la amiloidogénesis como lesión primaria).
Las proteínas Tau (predominan en los
axones) forman el grupo de las MAP (Microtubule Associated Protein) que
interactúan con los microtúbulos durante los movimientos y el transporte celular
ensamblando o desarmando los microtúbulos (acciones llamadas de rescate y
catástrofe respectivamente) en dependencia de si existe elongación o acortamiento
de las prolongaciones, en especial en los axones.
La hiperfosforilación de estas proteínas
provoca su precipitación y auto agregación formando, en el caso de la EA,
filamentos helicoidales pareados que entorpecen el transporte axonal con
neurodegeneración por posible apoptosis.
Los compuestos neurofibrilares son tan
insolubles y difíciles de proteilizar que aun después de la muerte neuronal,
permanecen como el vestigio o el esqueleto de aquella.
Un problema complejo es la
determinación de cuál es la lesión primaria y la relación entre ambas. Existen algunos
hechos que sugieren que lo primario es la acumulaciónBA. Por ejemplo, la ac umulación
BA precede a la presencia de ONF.
También se han logrado ratones K.O que
producen acumulación BA con todas las características de estas lesiones sin
degeneración neurofibrilar.
Dichos ratones presentan un déficit cognitivo
superponible a la EA humana.
Por otro lado, la demostración de la
proteína Tau como base de degeneraciones que cursan con demencia (ALS +
Alzheimer + Parkinson asociados al cromosoma 17, PSP y demencia de Pick) en ausencia
de PS, apoyan la segunda teoría.9,10
Algunos investigadores sugieren que
ambas lesiones son sólo cicatrices de un evento primario aún no descrito.
La posible relación entre las PS y los
ONF se postula en el daño que ocasionan los fragmentos BA insolubles en su paso
intracelular. Se cree que la respuesta inflamatoria secundaria pudiera dañar el
metabolismo de la proteína Tau.
Dicha relación no está completamente
probada y en contra de su existencia están la presencia de unas lesiones sin
las otras (en diferentes patologías), así como la diferente distribución de las
lesiones en un mismo cerebro.
Otros cambios que pudieran tener algún
peso en la pérdida neuronal son los del metabolismo del Ca (cascada enzimática),
el desequilibrio de los radicales libres, la toxicidad de algunos elementos
como el aluminio.
Los factores vasculares están recibiendo mayor atención debido al efecto
regulador de los núcleos colinérgicos sobre el flujo vascular regional. Otros factores
citados en la hipótesis neurovascular son la senescencia del árbol vascular, la
angiogénesis aberrante y el fallo del aclaramiento de BA a través de la barrera
hematoencefálica.
.
Reacción inflamatoria microglial y glial
Alrededor de los focos de amiloide se
produce una acumulación de neuritas distróficas que contienen DNF, junto con
una reacción de células microgliales y astrocitarias activadas.
La patogenia de esta reacción celular
se relaciona con la probable secreción de citocinas. La reacción inflamatoria
puede ser un paso intermedio muy importante en el daño neuronal a través de la
secreción de citocinas y el estrés oxidativo, que activan la cascada de la
apoptosis.

Activación macrofágica
Dado que la EA está íntimamente asociada a la activación de macrófagos cerebrales residentes, se han evaluado los niveles de activación macrofágica en sangre periférica de enfermos de Alzheimer y el grado de apoptosis linfocítica. Se ha encontrado una correlación entre los fenómenos de activación macrofágica y apoptosis linfocitaria existente dependiendo del grado de la severidad clínica de la enfermedad. Se concluye entonces que el fenómeno de activación macrofágica periférica y apoptosis es un fenómeno paralelo al proceso cerebral inflamatorio en los enfermos de Alzheimer.
Pérdida sináptica y neuronal
Tanto los oligómeros de Aβ como la
proteína τ anormal producen disfunción sináptica en modelos experimentales. La
muerte neuronal se produce, probablemente, por activación de las diferentes
vías de la apoptosis. La DNF como anomalía del citoesqueleto tiene
consecuencias catastróficas para la supervivencia neuronal.
GENÉTICA
La EA es una entidad heterogénea, que
se presenta de forma familiar o esporádica. Las formas familiares son relativamente
infrecuentes, menos de 1% y tiene un patrón dominante (AD). En las formas
esporádicas existen antecedentes de demencia en más de 80% por lo que se
sugiere un fuerte componente genético como factor de riesgo.
Los hallazgos genéticos comenzaron con
el descubrimiento de la proteína precursora amiloide (APP) en el cerebro de
portadores de Síndrome de Down con deterioro cognitivo.Éste se encontraba en el
cromosoma 21 y tenía un patrón de herencia AD con inicio temprano.
El gen de APP codifica por armazón
alternativa. La forma más grande es un polipéptido de 770 aminoácidos. El empalme
alternativo del exón 7 (que codifica el dominio Kurnitz) y del exón 8 (que codifica
el antígeno ox-2) resulta en un polipéptido de 695 aminoácidos (predomina) o en
otro de 751.
Le seguirían los descubrimientos de
los genes de las presenilinas 1 (cromosoma 14) y 2 (cromosoma 1), ambas con
patrón AD.17,18
La presencia del gen de la
apolipoproteína épsilon (APOE; cromosoma 19) como factor de riesgo para la EA
de inicio tardío está demostrado.19 Este gen tiene varios alelos: el 2, el 3 y
el 4. Tanto en sujetos normales como en EA el menos frecuente es el 2 y el más
común el 3; sin embargo, en sujetos con EA se observa el alelo E4 con una
frecuencia casi igual al 3.
El gen APOE 4 se puede presentar tanto de
forma heterocigótica (inicio de la enfermedad entre 5 y 10 años antes) como
homocigoto (inicio de la enfermedad entre 10 y 20 años antes).
La APOE se produce predominantemente en
los astrocitos y se introduce en la neurona a través de los receptores LDL, una
vez dentro se une a los ovillos neurofibrilares.
Este receptor es la vía común de la
A2M, APOE4 y la LDL. El antecedente de familiar con EA aumenta el riesgo de 2 a
7 veces.
La presencia de mutaciones de la A2M
está siendo estudiada como posible factor de riesgo para la EA de inicio tardío
pues se encuentra en 30% de estos pacientes.
|
Genotipo
en la enfermedad de Alzheimer Edad de inicio Cromosoma Herencia Producto Frecuencia
(%) Temprano 28-50 años 14 AD Presenilina 1 < 1 40-50 años 1 AD Presenilina 2 1 45-65 años 21 AD APP < 1 Tarde o nunca 19 AR APOE 4 > 65 |
FISIOPATOLOGÍA
Actualmente
existen dos teorías que tratan de explicar los déficit cognitivos de la EA: Teoría
de desconexión cortical y Teoría colinérgica.
Teoría de
desconexión cortical
La
degeneración neurofibrilar en la corteza entorrinal, portal cortical del hipocampo
(HC), se distribuyen en las cortezas II (que junto a la capa III forman la vía
perforante hacia el HC) y IV (que recibe la eferencia desde el HC) de manera
que el HC queda aislado de la neocorteza.
Esto se une
al déficit de glutamato y otros neuropéptidos (neuropéptido Y, oxitocina, vasopresina
y somatostatina) en las cortezas de asociación (desconexión córtico-cortical) que
correlaciona con la afasia, la apraxia y la agnosia, así como con los
trastornos visuoespaciales y ejecutivos.
Teoría
colinérgica
En estados
avanzados se observa una disminución de más de 90% de la actividad de la
acetilcolinesterasa lo que identifica un compromiso dramático del sistema colinérgico
en esta enfermedad. Esto ocasiona el deterioro mnésico inicial y progresivo.
La degeneración selectiva del núcleo basal de Meynert (principal eferencia colinérgica hacia neocortex) y de los núcleos septal y de la banda diagonal de broca (eferencia colinérgica subcortical, en especial hacia el HC) provocan un déficit progresivo de la memoria anterógrada.
Existen evidencias de cambios
tempranos en el flujo cerebral regional, lo que pudiera relacionarse con la degeneración
en la población colinérgica que tiene un efecto regulador.
Esto se conoce como teoría colinérgico-vascular. Por otro lado los desequilibrios de otras vías neuroquímicas explican mejor los síntomas no cognitivos.
Como se mencionó existe una afectación
de los núcleos superiores del rafe, el núcleo cerúleo y una conservación relativa
de la sustancia nigra.
• Déficit de serotonina
Se relaciona con los síntomas depresivos
así como con obsesión, compulsión y agresividad. Esto se observa tanto en EA
como en personas normales.
• Déficit de noradrenalina
Se observa también asociada a la
depresión y a la agitación psicomotora. Con este neurotransmisor ocurre algo
singular pues a pesar de existir una depoblación del núcleo cerúleo (donde se
observan Cuerpos de Lewy), existe una hiperactividad noradrenérgica cortical,
lo cual se atribuye a un aumento de la sensibilidad cortical y a la producción
de noradrenalina (NA) en corteza.
El aumento de la sensibilidad se
observa tanto en la corteza prefrontal como en el HC. Sin embargo, el aumento de
la concentración de NA sólo se encuentra en el cortex prefrontal. En los casos de
depresión existe disminución de NA, mientras que en aquellos con agitación existe
un aumento de ésta.
• Déficit de acetilcolina.
Como ya fue descrito, se asocia al
deterioro cognitivo, especialmente con los problemas de memoria. Sin embargo,
se postula que para que se desarrolle la depresión debe existir indemnidad o
niveles de acetilcolina cercanos a la normalidad. Esto sólo ocurre en los
estadios iniciales.
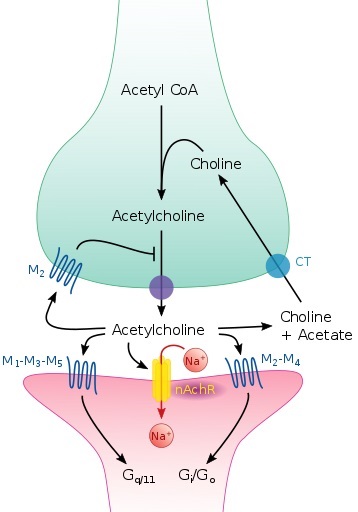
El principal defecto neuroquímico en
la EA se encuentra en el sistema colinérgico por la lesión del núcleo basal de
Meynert, que envía abundantes proyecciones colinérgicas a la corteza cerebral.
La actividad de la
colinoacetiltransferasa, enzima limitante de la síntesis de acetilcolina a
partir de colina y acetilcoenzima
A, está muy reducida en la neocorteza.
Además, los estudios neuroquímicos han demostrado un déficit en casi todos los
neurotransmisores clásicos y también en varios sistemas peptidérgicos
• Conservación relativa de dopamina.:
Este hecho provoca un desequilibrio
colina/dopamina con el aumento relativo de esta última observándose
alucinaciones, trastornos del sueño y psicosis. En un 30% existe un déficit de dopamina
con la aparición de un síndrome parkinsoniano. Sin embargo la preservación de
la postura y la marcha hasta estadios avanzados es una característica de las
demencias corticales por lo que los pacientes deambulan sin fin.
Estadios según Braak en la enfermedad de Alzheimer
Braak y Braak describieron 6 estadios
en la neuropatología de la EA . Las células de proyección específica de la
región transentorrinall perialocortical, que se localizan en las profundidades
del surco rinal, son las primeras neuronas corticales que manifiestan los
cambios (estadio I clínicamente silencioso).
Los casos de estadio II muestran
numerosos ovillos neurofibrilares y hilos del neurópilo en la región
transentorrinall, y algunos adicionales en la región entorrinal. La destrucción
cortical en el estadio II apenas impide la transmisión de la información
neocortical (a través de la región entorrinal) a la formación hipocampal, pero
sin exceder del umbral sobre el cual aparecen los síntomas clínicos iniciales.
Algunos individuos desarrollan ONs/HNs
iniciales a una edad sorprendentemente joven. Obviamente, la edad avanzada no
es un prerrequisito para el desarrollo de la patología intraneuronal. Esta
obsevación pone en duda teorías que intentan explicar los cambios como consecuencia
de influencias nocivas que se espera que tengan efecto generalmente en la edad
avanzada (estrés peroxidativo, disfunción mitocondrial, desequilibrio del
metabolismo de la glucosa). Esto no excluye la posibilidad de que el estrés
peroxidativo pueda contribuir a los cambios en los estadios avanzados de la
enfermedad o influencie en el proceso patológico. Posiblemente no parece ser un
factor primario en la patogénesis de las lesiones iniciales en la EA.
En los casos de estadios límbicos III o IV, la destrucción cortical es ya severa, pero limitada a unas pocas regiones alocorticales y áreas adyacentes. La característica clave de estos estadios es la notable destrucción de las capas entorrinals responsables de la transmisión de datos desde la neocorteza hasta el hipocampo y viceversa. Inicialmente, la formación hipocampal se ve afectada sólo ligeramente.
En el estadio IV el
proceso de destrucción se difunde desde la región entorrinal hacia la amígdala,
el hipocampo y especialmente hacia las áreas de asociación de la neocorteza
temporal basal. Los protocolos clínicos de muchos individuos en el estadio III
o IV registran un deterioro de las funciones cognitivas y presencia de cambios
sutiles de la personalidad. En otros individuos, la aparición de síntomas está
todavía camuflada por capacidades de reserva del individuo, las cuales
compensan la destrucción local. A causa de la expresión ocasional de los
síntomas iniciales y las lesiones cerebrales características, los casos de
estadios III y IV, se considera que representan la EA incipiente.
Los estadios neocorticales finales
muestran una gran cantidad de ONs y HNs en cada subdivisión de la corteza
cerebral. Una característica propia del estadio V es la destrucción
extremamente severa de las áreas asociativas neocorticales, dejando solamente
poco o nada afectados a los campos motores primarios, las áreas sensoriales
primarias y sus regiones circundantes.
En el estadio VI, el proceso
patológico se extiende a las áreas primarias. Los individuos caracterizados en
los estadios V y VI de destrucción cerebral son todos dementes. Estos estadios
corresponden a la EA completamente desarrollada.






Comentarios
Publicar un comentario